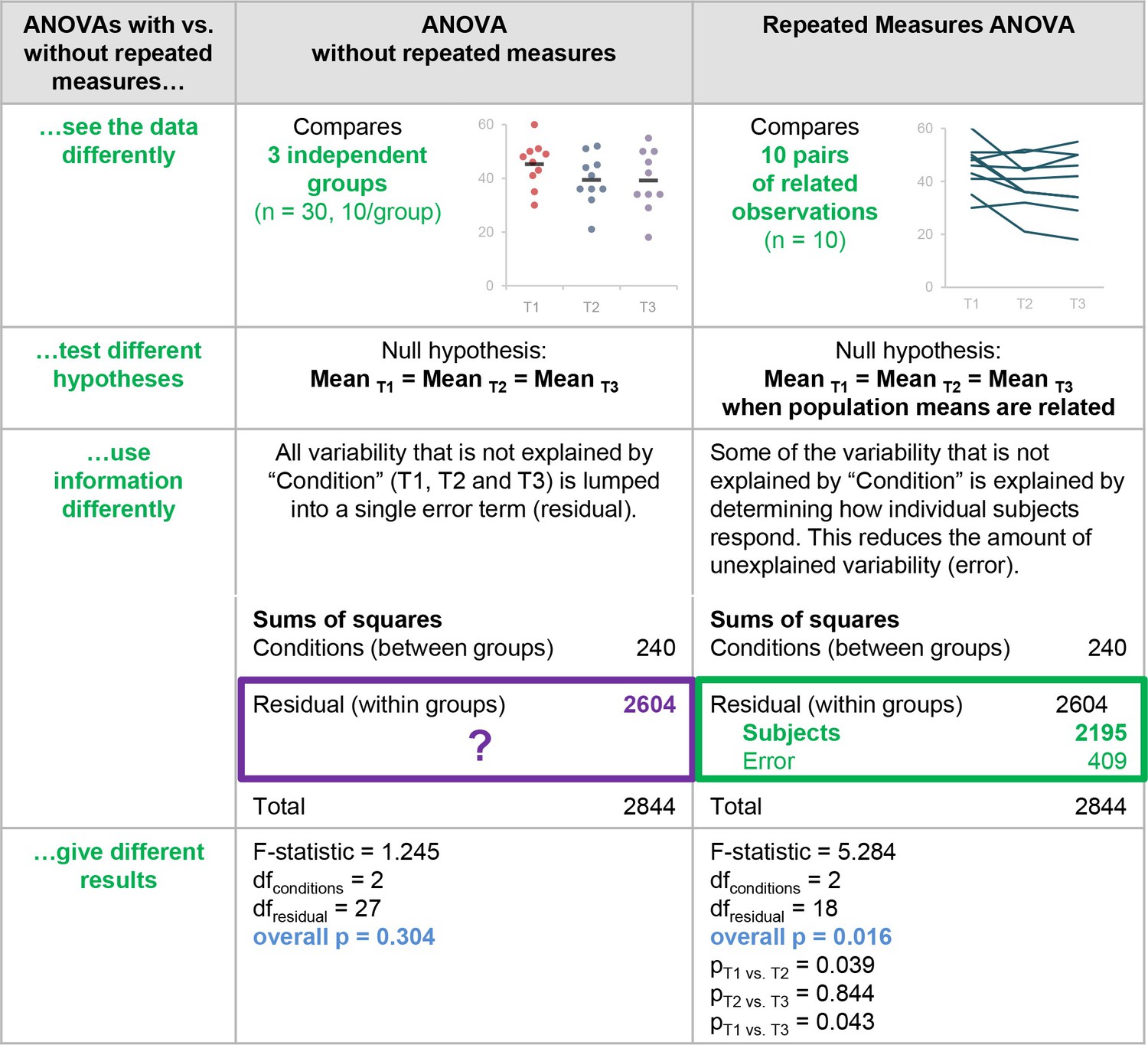
You want to test whether two samples are drawn from populations with different means, or test whether one sample is drawn from a population with a mean different from some theoretical mean. t.test(x, y = NULL, alternative = c(“two.sided”, “less”, “greater”), mu = 0, paired = FALSE (paired t-test), var.equal = FALSE (uses the Welch t-test or Student’s t-test), conf.level = 0.95, …)
Data which show the effect of two soporific drugs (increase in hours of sleep compared to control) on 10 patients.
extra: increase in hours of sleep group: drug given ID : patient ID
##
## Two Sample t-test
##
## data: extra by group
## t = -1.8608, df = 18, p-value = 0.07919
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -3.363874 0.203874
## sample estimates:
## mean in group 1 mean in group 2
## 0.75 2.33
# Not too significant indeed
Paired t-test
You might have observations before and after a treatment, or of two matched subjects with different treatments.
# with(sleep,# t.test(extra[group == 1],# extra[group == 2], paired = TRUE)) # t.test(extra ~ group, data = sleep,paired=TRUE)# with(sleep,# t.test(extra[group == 1] - extra[group == 2], mu=0, var.equal=TRUE)# )# Paired t-test/One Sample t-test# # data: extra by group# t = -4.0621, df = 9, p-value = 0.002833# alternative hypothesis: true difference in means is not equal to 0# 95 percent confidence interval:# -2.4598858 -0.7001142# sample estimates:# mean of the differences # -1.58 with(sleep,
stripchart(extra[group ==2] -extra[group ==1], method ="stack", xlab ="hours",
main ="Sleep prolongation (n = 10)")
)
with(sleep,
boxplot(extra[group ==2] -extra[group ==1], horizontal =TRUE, add =TRUE,
at = .6, pars =list(boxwex =0.5, staplewex =0.25))
)
MISC
Comparing a group against an expected population mean: one-sample t-test
Suppose that you want to test whether the data in column extra is drawn from a population whose true mean is 0.
t.test(sleep$extra, mu=0)
##
## One Sample t-test
##
## data: sleep$extra
## t = 3.413, df = 19, p-value = 0.002918
## alternative hypothesis: true mean is not equal to 0
## 95 percent confidence interval:
## 0.5955845 2.4844155
## sample estimates:
## mean of x
## 1.54
Frequency tests
There are two general questions that frequency tests address:
Does the frequency distribution differ from an expected, or theoretical, distribution (e.g., expect 50% yes, 50% no)? (Goodness-of-fit test)
Does the frequency distribution differ between two or more groups? (Independence test)
Expected distribution
Comparing groups
Exact
Exact binomial
Fisher’s exact
Approximate
Chi-square goodness of fit
Chi-square test of independence
Note: The exact binomial test can only be used on one variable that has two levels. Fisher’s exact test can only be used with two-dimensional contingency tables (for example, it can be used when there is one independent variable and one dependent variable, but not when there are 2 IVs and 1 DV).
To test for paired or within-subjects effects, McNemar’s test can be used. To use it, there must be one IV with two levels, and one DV with two levels.
To test for the independence of two variables with repeated measurements, the Cochran-Mantel-Haenszel test can be used.
Chi-square test
# chisq.test(c(5,7))# data: c(5, 7)# X-squared = 0.33333, df = 1, p-value = 0.5637
M <-as.table(rbind(c(762, 327, 468), c(484, 239, 477)))
dimnames(M) <-list(gender =c("F", "M"),
party =c("Democrat","Independent", "Republican"))
# party# gender Democrat Independent Republican# F 762 327 468# M 484 239 477chisq.test(M,p=c(0.3,0.3,0.4)) # Pearson’s chi-square test
##
## Pearson's Chi-squared test
##
## data: M
## X-squared = 30.07, df = 2, p-value = 2.954e-07
exact binomial test
binom.test(c(682, 243), p =3/4)
##
## Exact binomial test
##
## data: c(682, 243)
## number of successes = 682, number of trials = 925, p-value =
## 0.3825
## alternative hypothesis: true probability of success is not equal to 0.75
## 95 percent confidence interval:
## 0.7076683 0.7654066
## sample estimates:
## probability of success
## 0.7372973
Fisher’s exact test
TeaTasting <-matrix(c(3, 1, 1, 3),nrow =2,
dimnames =list(Guess =c("Milk", "Tea"),
Truth =c("Milk", "Tea")))
fisher.test(TeaTasting, alternative ="greater")
##
## Fisher's Exact Test for Count Data
##
## data: TeaTasting
## p-value = 0.2429
## alternative hypothesis: true odds ratio is greater than 1
## 95 percent confidence interval:
## 0.3135693 Inf
## sample estimates:
## odds ratio
## 6.408309
Cochran-Mantel-Haenszel test
The Cochran-Mantel-Haenszel test (or Mantel-Haenszel test) is used for testing the independence of two dichotomous variables with repeated measurements. It is most commonly used with 2x2xK tables, where K is the number of measurement conditions. For example, you may want to know whether a treatment (C vs. D) affects the likelihood of recovery (yes or no). Suppose, though, that the treatments were administered at three different times of day, morning, afternoon, and night – and that you want your test to control for this. The CMH test would then operate on a 2x2x3 contingency table, where the third variable is the one you wish to control for.
The implementation of the CMH test in R can handle dimensions greater than 2x2xK. For example, you could use it for a 3x3xK contingency table.
fish <-read.table(header=TRUE, text=' Location Allele Habitat Count tillamook 94 marine 56 tillamook 94 estuarine 69 tillamook non-94 marine 40 tillamook non-94 estuarine 77 yaquina 94 marine 61 yaquina 94 estuarine 257 yaquina non-94 marine 57 yaquina non-94 estuarine 301 alsea 94 marine 73 alsea 94 estuarine 65 alsea non-94 marine 71 alsea non-94 estuarine 79 umpqua 94 marine 71 umpqua 94 estuarine 48 umpqua non-94 marine 55 umpqua non-94 estuarine 48')
# Make a 3D contingency table, where the last variable, Location, is the one to # control for. If you use table() for case data, the last variable is also the # one to control for.
ct <-xtabs(Count ~Allele +Habitat +Location, data=fish)
ct
##
## Mantel-Haenszel chi-squared test with continuity correction
##
## data: ct
## Mantel-Haenszel X-squared = 5.0497, df = 1, p-value = 0.02463
## alternative hypothesis: true common odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.6005522 0.9593077
## sample estimates:
## common odds ratio
## 0.759022
According to this test, there is a relationship between Allele and Habitat, controlling for Location, p=.025.
Note that the first two dimensions of the contingency table are treated the same (so their order can be swapped without affecting the test result), the highest-order dimension in the contingency table is different. This is illustrated below.
# The following two create different contingency tables, but have the same result# with the CMH test
ct.1 <-xtabs(Count ~Habitat +Allele +Location, data=fish)
ct.2 <-xtabs(Count ~Allele +Habitat +Location, data=fish)
mantelhaen.test(ct.1)
##
## Mantel-Haenszel chi-squared test with continuity correction
##
## data: ct.1
## Mantel-Haenszel X-squared = 5.0497, df = 1, p-value = 0.02463
## alternative hypothesis: true common odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.6005522 0.9593077
## sample estimates:
## common odds ratio
## 0.759022
mantelhaen.test(ct.2)
##
## Mantel-Haenszel chi-squared test with continuity correction
##
## data: ct.2
## Mantel-Haenszel X-squared = 5.0497, df = 1, p-value = 0.02463
## alternative hypothesis: true common odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.6005522 0.9593077
## sample estimates:
## common odds ratio
## 0.759022
# With Allele last, we get a different result
ct.3 <-xtabs(Count ~Location +Habitat +Allele, data=fish)
ct.4 <-xtabs(Count ~Habitat +Location +Allele, data=fish)
mantelhaen.test(ct.3)
# With Habitat last, we get a different result
ct.5 <-xtabs(Count ~Allele +Location +Habitat, data=fish)
ct.6 <-xtabs(Count ~Location +Allele +Habitat, data=fish)
mantelhaen.test(ct.5)
McNemar’s test is conceptually like a within-subjects test for frequency data. For example, suppose you want to test whether a treatment increases the probability that a person will respond “yes” to a question, and that you get just one pre-treatment and one post-treatment response per person. A standard chi-square test would be inappropriate, because it assumes that the groups are independent. Instead, McNemar’s test can be used. This test can only be used when there are two measurements of a dichotomous variable. The 2x2 contingency table used for McNemar’s test bears a superficial resemblance to those used for “normal” chi-square tests, but it is different in structure.
data <-read.table(header=TRUE, text=' subject time result 1 pre 0 1 post 1 2 pre 1 2 post 1 3 pre 0 3 post 1 4 pre 1 4 post 0 5 pre 1 5 post 1 6 pre 0 6 post 1 7 pre 0 7 post 1 8 pre 0 8 post 1 9 pre 0 9 post 1 10 pre 1 10 post 1 11 pre 0 11 post 0 12 pre 1 12 post 1 13 pre 0 13 post 1 14 pre 0 14 post 0 15 pre 0 15 post 1')
library(tidyr)
data_wide <-spread(data, time, result)
ct <-table( data_wide[,c("pre","post")] )
ct
## post
## pre 0 1
## 0 2 8
## 1 1 4
#> post#> pre 0 1#> 0 2 8#> 1 1 4# The contingency table above puts each subject into one of four cells, depending# on their pre- and post-treatment response. Note that it it is different from# the contingency table used for a "normal" chi-square test, shown below. The table# below does not account for repeated measurements, and so it is not useful for# the purposes here.# table(data[,c("time","result")])# result# time 0 1# post 3 12# pre 10 5mcnemar.test(ct)
For small sample sizes, it uses a continuity correction. Instead of using this correction, you can use an exact version of McNemar’s test, which is more accurate. It is available in the package exact2x2.
##
## Exact McNemar test (with central confidence intervals)
##
## data: ct
## b = 8, c = 1, p-value = 0.03906
## alternative hypothesis: true odds ratio is not equal to 1
## 95 percent confidence interval:
## 1.072554 354.981246
## sample estimates:
## odds ratio
## 8
Homogeneity of variance
You want test samples to see for homogeneity of variance (homoscedasticity) – or more accurately. Many statistical tests assume that the populations are homoscedastic. - Bartlett’s test - If the data is normally distributed, this is the best test to use. It is sensitive to data which is not non-normally distribution; it is more likely to return a “false positive” when the data is non-normal. - Levene’s test - this is more robust to departures from normality than Bartlett’s test. It is in the car package. - Fligner-Killeen test - this is a non-parametric test which is very robust against departures from normality.
For all these tests, the null hypothesis is that all populations variances are equal; the alternative hypothesis is that at least two of them differ.
head(InsectSprays)
## count spray
## 1 10 A
## 2 7 A
## 3 20 A
## 4 14 A
## 5 14 A
## 6 12 A
tg <-ToothGrowth
tg$dose <-factor(tg$dose) # Treat this column as a factor, not numerichead(tg)
# On a first glance, it appears that both data sets are heteroscedastic, but this needs to be properly tested, which we’ll do below.
Bartlett’s test
With one independent variable:
bartlett.test(count ~spray, data=InsectSprays)
##
## Bartlett test of homogeneity of variances
##
## data: count by spray
## Bartlett's K-squared = 25.96, df = 5, p-value = 9.085e-05
With multiple independent variables, the interaction() function must be used to collapse the IV’s into a single variable with all combinations of the factors. If it is not used, then the will be the wrong degrees of freedom, and the p-value will be wrong.
## Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 5 3.8214 0.004223 **
## 66
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
With two independent variables. Note that the interaction function is not needed, as it is for the other two tests.
leveneTest(len ~supp*dose, data=tg)
## Levene's Test for Homogeneity of Variance (center = median)
## Df F value Pr(>F)
## group 5 1.7086 0.1484
## 54
Fligner-Killeen test
# With one independent variable:fligner.test(count ~spray, data=InsectSprays)
##
## Fligner-Killeen test of homogeneity of variances
##
## data: count by spray
## Fligner-Killeen:med chi-squared = 14.483, df = 5, p-value =
## 0.01282
The fligner.test function has the same quirks as bartlett.test when working with multiple IV’s. With multiple independent variables, the interaction() function must be used.
If the data is ordinal, then it may be appropriate to use a weighted Kappa. For example, if the possible values are low, medium, and high, then if a case were rated medium and high by the two coders, they would be in better agreement than if the ratings were low and high.
We will use a subset of the anxiety data set from the irr package.
library(irr)
data(anxiety)
dfa <-anxiety[,c(1,2)]
The weighted Kappa calculation must be made with 2 raters, and can use either linear or squared weights of the differences.
# Compare raters 1 and 2 with squared weightskappa2(dfa, "squared")
The data above is numeric, but a weighted Kappa can also be calculated for factors. Note that the factor levels must be in the correct order, or results will be wrong.
# Make a factor-ized version of the data
dfa2 <-dfa
dfa2$rater1 <-factor(dfa2$rater1, levels=1:6, labels=LETTERS[1:6])
dfa2$rater2 <-factor(dfa2$rater2, levels=1:6, labels=LETTERS[1:6])
dfa2
## rater1 rater2
## 1 C C
## 2 C F
## 3 C D
## 4 D F
## 5 E B
## 6 E D
## 7 B B
## 8 C D
## 9 E C
## 10 B C
## 11 B B
## 12 F C
## 13 A C
## 14 E C
## 15 B B
## 16 B B
## 17 A A
## 18 B C
## 19 D C
## 20 C D
# The factor levels must be in the correct order:levels(dfa2$rater1)
## [1] "A" "B" "C" "D" "E" "F"
#> [1] "A" "B" "C" "D" "E" "F"levels(dfa2$rater2)
## [1] "A" "B" "C" "D" "E" "F"
#> [1] "A" "B" "C" "D" "E" "F"# The results are the same as with the numeric data, abovekappa2(dfa2, "squared")
When the variable is continuous, the intraclass correlation coefficient should be computed. From the documentation for icc:
When considering which form of ICC is appropriate for an actual set of data, one has take several decisions (Shrout & Fleiss, 1979):
Should only the subjects be considered as random effects ("oneway" model, default) or are subjects and raters randomly chosen from a bigger pool of persons ("twoway" model).
If differences in judges’ mean ratings are of interest, interrater "agreement" instead of "consistency" (default) should be computed.
If the unit of analysis is a mean of several ratings, unit should be changed to "average". In most cases, however, single values (unit="single", default) are regarded.
We will use the anxiety data set from the irr package.